Маркеры дефектов хромосом
Содержание:
Также в разделе
|
Синдром множественных птеригиумов Данный синдром (имеется в виду нелетальная форма) является самой известной нозологической единицей среди синдромов-множественных птеригиумов. Из… |
|
| Делеция дистального отдела длинного плеча хромосомы 11 Делеция дистального отдела длинного плеча хромосомы 11 впервые описана P. Jacobsen, спустя 2 года стали говорить о клиническом синдроме, сопровождающем такую… | |
| Врожденные пороки прямой кишки и анального отверстия По данным разных авторов, аноректальные аномалии встречаются с частотой 0,25-0,66 случая на 1000 новорожденных; у девочек в 2 раза реже, чем у мальчиков. Характерной… | |
| Морфологические изменения при патологии развития, захватывающей весь эмбрион В первые 6-7 нед беременности нередко встречаются морфогенетические изменения, захватывающие весь эмбрион, вызывающие общие тяжелые нарушения развития и… | |
|
Врожденные пороки пищевода Агенезия пищевода у новорожденных встречается крайне редко и сочетается с другими тяжелыми нарушениями развития. Гипоплазия проявляется укорочением… |
|
| Пороки развития вентрикулярной системы и подпаутинного (субарахноидального) пространства Пороки развития вентрикулярной системы обычно возникают в области ее анатомических сужений: межжелудочковых отверстий водопровода среднего мозга, срединной… | |
| Системные врожденные заболевания скелета Системные (генерализованные) врожденные заболевания или диспластическне поражения костей составляют обширную и гетерогенную группу аномалий, в основе… | |
| Ассоциация CHARGE Об ассоциации CHARGE говорят с 1981 г., когда на основании анализа предшествующей литературы и 21 наблюдения отметили необычно частое сочетание между собой колобом… | |
| Делеция длинного плеча хромосомы 18 Для больных с делецией длинных плеч I8q- характерны микроцефалия, гипертелоризм, дизморфия лица (уплощение спинки маленького по размерам носа, глубоко… | |
| Врожденные пороки толстой кишки При аплазии слепой кишки и червеобразного отростка непрерывность желудочно-кишечного тракта обычно сохранена, но характерное расширение кишечной трубки, в… |
Лечение[править | править код]
Пациенты обычно должны наблюдаться у эндокринолога. При наличии гипогонадизма лечение тестостероном следует рассматривать у всех людей, независимо от когнитивных способностей, из-за положительного влияния на здоровье костей, мышечный тонус, усталость и выносливость, а также с возможными преимуществами для психического здоровья / поведения.
У большинства детей с XXYY наблюдаются некоторые задержки в развитии и трудности в обучении. Следовательно, эти аспекты должны наблюдаться: психология (когнитивное и социально-эмоциональное развитие), логопедия, трудотерапия и физиотерапия. Следует организовать консультации с развивающими педиатрами, психиатрами или неврологами для разработки плана лечения, включающего терапию, поведенческие вмешательства, образовательную поддержку и психотропные препараты для поведенческих и психиатрических симптомов. Общие диагнозы, такие как неспособность к обучению, СДВГ, расстройства аутистического спектра, перепады настроения, тиковые расстройства и другие проблемы психического здоровья, должны быть рассмотрены, обследованы и пролечены. В этой группе наблюдаются хорошие реакции на стандартные медикаментозные методы лечения от невнимательности, импульсивности, тревоги и нестабильности настроения, и такое лечение может положительно влиять на успеваемость, эмоциональное благополучие в долгосрочной перспективе. Плохая координация мелкой моторики может сделать письмо медленным и трудоемким занятием, и трудотерапия и клавиатура должны быть введены в раннем возрасте, чтобы облегчить школьную работу и навыки самопомощи. Образовательные трудности должны оцениваться с помощью полной психологической оценки, чтобы выявить расхождения между устными и служебными навыками и выявить индивидуальные академические потребности. Языковые навыки часто страдают в течение всей жизни, и в зрелом возрасте могут потребоваться вмешательства в области логопедической терапии, направленные на развитие выразительных языковых навыков, диспраксии и др. Адаптивные навыки представляют собой затруднения, требующие поддержки на уровне сообщества почти для всех людей в зрелом возрасте. Могут потребоваться дополнительные рекомендации по лечению, основанные на индивидуальных сильных и слабых сторонах синдрома XXYY.
Показания к исследованию
Исходя из описания методов исследования, уже можно предположить, что генетические заболевания встречаются не так уж и редко. По статистике ВОЗ, различные пороки развития встречаются примерно у 3% детей, а выявляемость их к семилетнему возрасту возрастает с частотой по проценту в год, до 7%. Конечно, речь идет о крупных городах – миллионниках, где есть специалисты – генетики и высокотехнологические лаборатории. Следует учесть, что ни один из современных секвенсоров нового поколения не производится в РФ, оборудование стоит очень дорого.
Пренатальный скрининг и беременные
Основным показанием к проведению генетических исследований являются нарушения и подозрения в сфере репродуктологии. К ним относят:
- различные варианты бесплодия, тем более повторного;
- случаи внутриутробной смерти плода;
- повторные выкидыши у одной и той же женщины;
- наличие у братьев и сестёр наследственных заболеваний, или значимых признаков;
- необъяснимое отставание в развитии.
Кроме пренатальной диагностики, генетические исследования обязательно проводятся в период беременности, и поводом для назначения анализа обычно является так называемый тройной тест, при котором выявляются уровни ХГЧ, эстриола и альфа-фетопротеина. На основании данных этого анализа проводится генетическое исследование, которое позволяет выявить опасные дефекты нервной трубки, синдромы Эдвардса, Патау, миопатию Дюшенна и другие болезни
Определение родства
Также очень важным и юридически значимым будет определение по наследственному анализу крови отцовства и материнства, этот анализ довольно часто фигурирует в различных судебных делах о неуплате алиментов и по разделе наследства, а также при поиске наследников. Средняя стоимость такого анализа – около 25 тысяч рублей.
Выявление симптомов у взрослых
Иногда бывает, что врач подозревает у человека генетические отклонения, связанные с некоторыми аномалиями развития или функции. Так, бесплодие, неправильное развитие половых органов, чрезмерно маленький или высокий рост, умственная отсталость, патологическая склонность к жестокости также может натолкнуть врача на необходимость проведения генетического исследования.
Модные направления
Современная концепция здорового образа жизни, взаимодействие с природой, стратегия прогнозирования рисков, вместо того чтобы лечить уже сформированные заболевания, привела к тому, что в генетических исследованиях стали превалировать профилактические направления. Это, прежде всего генетика предрасположенностей. В данном случае выявляются те особенности наследственного статуса пациента, которые могут стать решающими при воздействии определённых факторов внешней среды. Выявляется не заболевание, а наличие предрасположенности к ним, которая имеет повышенный риск реализации. Это диабет, онкологические заболевания, бронхиальная астма.
Трудно даже представить, какое разнообразие генетических анализов можно выполнить, если речь идет о предрасположенностях. Это, например, риск развития рака при курении, риск возникновения рака шейки матки, панель риска возникновения нарушений системы гемостаза (свертывания крови), в которой определяется сразу 30 маркёров риска, панель «высокое артериальное давление» и подбора лекарств.
Кстати, фармакогенетика является одной из бурно развивающихся видов генной диагностики. Она позволяет установить, нет ли наследственных особенностей, которые делают какой-либо назначенный пациенту препарат малоэффективным (вследствие низкой экспрессии генов ферментов печени, например, модифицирующих структуру пролекарства).
Существуют и такие анализы (нутригенетика) которые подсказывают пациенту на основании генетического анализа крови, какая диета будет оптимальной для снижения веса: низкожировая, низкоуглеводная или средиземноморская. Это исследование строится на основании анализа различных генов, которые кодируют синтез инсулина, переносчика жирных кислот, гены белков, ассоциированных с ожирением и быстрым набором веса. После анализа степени экспрессии каждого из этих генов делается вывод о том, какой из них сильнее, а у какого гена экспрессия не выражена, и на какой диете пациент будет худеть больше. Нутригенетика также рассматривает, какие витамины могут меньше усваиваться, какие тренировки нужны при данном типе генов – интенсивные, «спринтерские», или «стайерские» для оптимального набора мышечной массы.
История открытия синдрома
Синдром получил свое название в честь Гарри Клайнфельтера — врача, в 1942 году впервые описавшего клиническую картину болезни. Клайнфельтер с коллегами опубликовали отчет об обследовании 9 мужчин, объединенных общими симптомами, такими как слабое оволосение тела, евнухоидный тип телосложения, высокий рост и уменьшенные в размерах яички. Позднее, в 1956 г., генетики Планкетт и Барр (Е. R. Plankett, М. L. Barr) обнаружили у мужчин с синдромом Клайнфельтера тельца полового хроматина в ядрах клеток слизистой оболочки полости рта, а в 1959 году Полани и Форд (P. E. Polanyi, S. E. Ford) с сотрудниками показали, что у больных в хромосомном наборе имеется лишняя Х-хромосома.
Активные исследования данной патологии велись в 70‑х годах в США. Тогда всех новорожденных мальчиков подвергали кариотипированию, в результате чего удалось достоверно выявить распространенность и генетические особенности синдрома Клайнфельтера.
Любопытно, что мыши также могут иметь синдром трисомии по половым хромосомам XXY, что позволяет эффективно использовать их в качестве моделей для исследования синдрома Клайнфельтера.
Аномалии половых хромосом
Наиболее частыми аномалиями половых хромосом является синдром Тернера (моносомия Х, или 45, Х0, мозаицизм) и синдром Кляйнфельтера (47, ХХУ). Это может быть обусловлено тем обстоятельством, что кариотипы 47, ХХХ и 47, ХVV не имеют выраженных фенотипических различий и поэтому идентифицируются реже.
Синдром Тернера (Шерешевского — Тернера) может быть вызван потерей родительской хромосомы, мозаицизмом (45, Х / 46, ХХ или 45, Х / 46, ХУ) или структурными аномалиями Х-хромосомы (делеции, изохромосомы).
Индивиды с синдромом Тернера имеют женский фенотип, дисгенезию гонад, низкий рост, первичные, отсутствие вторичных половых признаков, крыловидные складки шеи, низко расположенные уши, низкую заднюю границу роста волос, дискообразную грудную клетку с широкой расстоянием между сосками, аномалии почек, лимфедему конечностей при рождении и кардиоваскулярные аномалии, чаще коарктацию аорты. Но при ультразвуковом исследовании может проявляться только одна аномалия — кистозная гигрома. Скрининг-теста синдромом Тернера еще не существует, и поэтому частота рецидивов не определена.
В случае синдрома Кляйнфельтера развитие яичек сначала является нормальным. Но присутствие не менее 1 лишней Х-хромосомы приводит к гибели зародышевых клеток на этапе их поступления в мейоз, что приводит к уменьшению яичек и гиалинизации семенных протоков. Итак, классические симптомы синдрома Кляйнфельтера включают бесплодие, гинекомастию, задержку умственного развития, повышения уровня гонадотропинов вследствие уменьшения уровня циркулирующих андрогенов. Скрининг-теста по выявлению синдрома Кляйнфельтера также не существует, следовательно, пренатальный диагноз этих хромосомных аномалий возможно только при использовании биопсии хориона или амниоцентеза.
Симптомы андрогенной недостаточности при синдроме Клайнфельтера
Андрогенная недостаточность при синдроме Клайнфельтера связана с постепенной атрофией яичек, что приводит к снижению синтеза тестостерона. Степень недостаточности андрогенов резко варьирует.
В первую очередь обращают на себя внимание внешние признаки гипогонадизма:
- скудная растительность на лице или же полное ее отсутствие;
- рост волос на лобке по женскому типу;
- волосы на груди и других частях тела отсутствуют;
- маленький объем яичек (2–4 мл) и их плотная консистенция (патогномоничный признак).
Поскольку дегенерация половых желез, как правило, развивается в постпубертатный период, у большинства пациентов размеры мужских половых органов, за исключением яичек, соответствуют возрастным нормам.
Пациенты могут жаловаться на ослабление либидо и снижение потенции. У многих мужчин с синдромом Клайнфельтера половое влечение вовсе не возникает, а некоторые — напротив, заводят семью и живут нормальной половой жизнью. Наиболее постоянный признак патологии — бесплодие, именно оно чаще всего становится причиной обращения таких пациентов к врачу. У 10 % мужчин с азооспемией обнаруживают синдром Клайнфельтера.
Всем пациентам с нарушениями сперматогенеза необходимо определять кариотип для исключения или подтверждения диагноза синдрома Клайнфельтера.
Недостаток андрогенов приводит к развитию остеопороза, анемии и слабости скелетной мускулатуры. У трети больных можно наблюдать варикозное расширение вен голеней.
Андрогены влияют на обмен веществ, поэтому больные с синдромом Клайнфельтера склонны к ожирению, нарушению толерантности к глюкозе и сахарному диабету второго типа.
Доказана предрасположенность таких пациентов к аутоиммунным заболеваниям (ревматоидный артрит, системная красная волчанка, аутоиммунные заболевания щитовидной железы и другие).
Скрининг на генетические заболевания
Сегодня известно более 11 000 моногенных заболеваний, которые кодируются одним геном (генетически обусловленные) и передаются от родителей их потомкам. Механизм передачи многих генетических болезней объясняется принципами Менделя.
Аутосомно-доминантные моногенные синдромы встречаются с частотой 1: 200 индивидов; заболевание наблюдается у многих поколений, передается потомкам и рецидивирует с частотой 50%. Примерами аутосомно-доминантных моногенных расстройств могут быть:
- ахондроплазия,
- нейрофиброматоз,
- синдром Марфана,
- болезнь Хантингтона,
- семейный полипоз.
Появление аутосомно-доминантных заболеваний у новорожденных от «здоровых» родителей может быть обусловлено следующими причинами:
1. Мозаицизм зародышевых клеток. Мутация может иметь место лишь в популяции зародышевых клеток. Итак, родители являются непораженными, но могут передавать мутацию потомкам.
2. Новые мутации. Рост возраста родителей ассоциируется с увеличением риска аутосомно-доминантных расстройств (ахондроплазии, танатофорной дисплазии, нейрофиброматоза, синдрома Аперта — краниосиностоз). Риск рецидивов у других детей не увеличивается.
3. Вариабельна экспрессия. Тяжесть заболевания может варьировать, и родители могут не распознать мягкие и субклинические мутации.
4. Уменьшенная пенетрантность. Родители могут иметь аномальный ген без клинических проявлений заболевания.
5. Неверное отцовство. Частота неверного отцовства достигает 15%.
Аутосомно-рецессивные моногенные заболевания проявляются в многочисленных родственников при наличии двух пораженных аллелей. Если оба родителя являются носителями пораженного гена, риск заболевания у потомства равен 25% при каждой беременности. Аутосомно-рецессивные заболевания включают кистозный фиброз, серповидно-клеточную анемию, фенилкетонурию, болезнь Тея-Сакса, Канавана и др.
При Х-сцепленных рецессивных синдромах (гемофилия и др.) мать-носитель пораженного гена передает его своим сыновьям. Итак, 50% сыновей могут быть больными и 50% дочерей будут носителями этого гена. Редкие Х-доминантные синдромы могут передаваться от каждого родителя каждому ребенку подобно аутосомно-доминантных синдромов. Фенотип может сильно варьировать, что связано со смешанной пенетрантностью, лионизацией (гетерохроматизацией) Х-хромосомы (синдром ломкой Х-хромосомы) и геномным импринтингом.
Экспансия тринуклеотидных повторов. Некоторые гены содержат участки тройных повторов (например, ССС). Такие участки являются нестабильными и могут увеличиваться в следующих генерациях, этот феномен получил название антиципации. Количество повторений определяет степень поражения индивида. Экспансия тринуклеотидных повторов составляет основу многочисленных генетических расстройств, таких как синдром ломкой (фрагильной) Х-хромосомы, миотоническая дистрофия и болезнь Хантингтона.
Синдром ломкой (фрагильной) Х-хромосомы является наиболее частой причиной семейной задержки умственного развития. Пораженные мужчины имеют типичные черты: большие уши, выступающая челюсть, большие яички, аутичное поведение, легкая или умеренная умственная отсталость. Женщины обычно менее поражены в связи с инактивацией Х-хромосомы.
Ген ломкой Х-хромосомы локализуется в Х-хромосоме и имеет три нуклеотидные повтора (ССС). Нормальные индивиды имеют 6-50 повторов, непораженные носители женского пола могут иметь 50-200 повторов, которые могут распространяться на мейоза до полной мутации при наличии более 200 повторов. Если имеет место полная мутация, ген инактивируется путем метилирования; плод будет пораженным. Тяжесть заболевания зависит от степени Х-инактивации у женщин, степени метилирования и мозаицизма размера повторов.
Женщины-носители премутации имеют 50%-й риск передачи гена с экспансией. Мужчины с премутациею фенотипически являются нормальными, но все их дочери будут носителями премутации. В случае трансмиссии мужчинам количество повторов остается стабильным. Тест на ломку Х-хромосому выполняется с целью выявления количества повторов и степени метилирования.
3.Синдром Эдвардса
Частота
встречаемости 1: 7000. Различают 2 формы:
—
трисомная по хромосоме 18 (90 % случаев);
—
мозаичная (10 % случаев).
Фенотип:
задержка роста, множественные аномалии
развития: долихоцефалический череп
(преобладание продольных размеров
головы над поперечным) с выступающим
затылком, «птичий» профиль лица, короткие
и горизонтально расположенные глазные
щели; маленькие, деформированные низко
расположенные уши, избыточная кожа на
затылке; микростомия (узкая ротовая
щель); флексорное сгибание кисти с
наложением указательного пальца на
III, а V на IV; «стопа – качалка» (с провисающим
сводом и выступающей кзади пяткой);
синдактилия; врождённые пороки сердца
и крупных сосудов. Продолжительность
жизни резко снижена.
Потенциальные проблемы на генном уровне
Проблем на уровне генов может быть несколько. И все они рассматриваются отдельно, ведь имеют разную клиническую картину. Ниже представлены только те патологии, которые современная медицина может успешно вылечить после того, как родился больной ребенок:
- Моносомия — патология, которая отличается отсутствием гомологичной хромосомы.
- Анэуплоидия — нарушается число отдельных единиц.
- Трисомия — когда в клетке присутствует лишняя хромосома (также есть патология тетрасомия, когда лишних хромосом две).
- Полиплоидия — количество гаплоидных наборов больше, чем диплоидных.
Эти показания считаются отклонением от нормы и их можно определить еще во время внутриутробного развития. Если существует возможного того, что ребенок родится с серьезными проблемами, врачи часто рекомендуют беременной женщине сделать аборт. В противном случае женщина обрекает себя на жизнь с инвалидом, которому будет необходимо дополнительное воспитание.
Диагностика генетической патологии
пренатальная диагностикаПренатальная диагностика синдрома Дауна включает следующие исследования:
- анализ родословной;
- кариотипирование родителей;
- ультразвуковое исследование (УЗИ);
- исследование сывороточных маркеров;
- исследование ДНК плода.
Кариотипирование родителей
лимфоцитымитозтрисомиюПоказанием для проведения данного исследования могут быть:
- возраст матери (во многих странах анализ назначают в обязательном порядке после 35 лет);
- трудности с зачатием ребенка в прошлом (выкидыши, внутриутробная смерть плода, и др.);
- наличие генетических заболеваний в роду у одного из супругов (по результатам анализа родословной);
- место и условия жизни супругов (области с повышенным радиоактивным фоном);
- неблагоприятные условия работы (воздействие сильного электромагнитного излучения, контакт с некоторыми химикатами);
- длительные перебои менструального цикла у женщины и некоторые гормональные заболевания;
- кровное родство с мужем (двоюродный/троюродный брат и т.п.);
- употребление в прошлом наркотических препаратов (оно могло повредить генетический материал в яйцеклетках, что повышает риск, даже если женщина уже много лет как излечилась от зависимости).
Ультразвуковое исследование
В первом триместре беременности при синдроме Дауна на УЗИ можно выявить следующие признаки патологии:
- утолщение воротникового пространства;
- шейная гигрома;
- отсутствие носовой кости;
- отставание плода в росте и весе от нормы на 8 – 10%.
Во втором триместре беременности удается выявить на УЗИ следующие признаки болезни:
- брахицефалия;
- увеличение объема сердечных желудочков;
- кисты в области сосудистых сплетений;
- киста в задней черепной ямке;
- недоразвитие костей лицевого черепа;
- наличие дополнительной складки на шее;
- неправильная форма большой мозговой цистерны;
- непроходимость кишечника (часто в области двенадцатиперстной кишки);
- пороки сердца различной степени тяжести;
- короткие трубчатые кости конечностей;
- аномалии развития пальцев;
- гидронефроз почек.
Исследование сывороточных маркеров
низкое разрешение аппарата, низкая квалификация врача, отсутствие видимых аномалийО наличии синдрома Дауна у плода могут говорить следующие маркеры:
- хорионический гонадотропин человека (ХГЧ);
- плазменный протеин А;
- эстриол;
- альфа-фетопротеин.
ХГЧ
Исследование ДНК плода
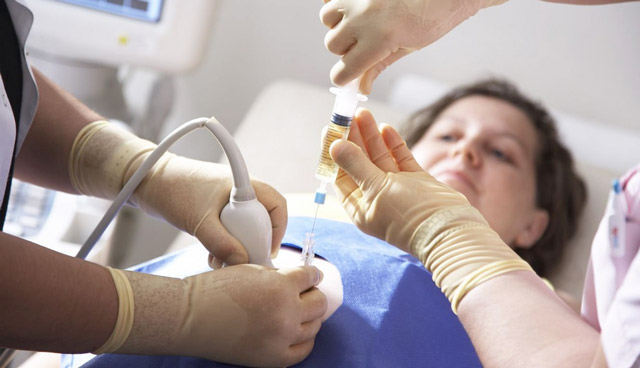
представляют собой довольно сложную процедуруК инвазивным методам получения генетического материала плода относятся:
- Кордоцентез. С помощью специальной тонкой иглы делается прокол в передней брюшной стенке матери. Игла вводится в сосуд пупочного канатика, и делается забор крови плода.
- Амниоцентез. Данный метод схож с кордоцентезом, но игла вводится не в сосуды пуповины, а в плодный мешок. Отсюда берут некоторое количество плодной жидкости, которая содержит клетки с поверхности кожи зародыша.
- Биопсия хориона. Техника выполнения аналогична предыдущим исследованиям. С помощью пункции делается забор ворсинок хориона (оболочки плода), которые также содержат ДНК будущего ребенка.
также именуемая ДОТ-тестПодтверждение диагноза после первичного осмотра новорожденного делается по следующим критериям:
- «уплощенное» лицо;
- отсутствие рефлекса Моро (в норме при ударе по поверхности рядом с ребенком он разводит руки в стороны и раскрывает ладони на несколько секунд);
- характерный разрез глаз;
- слабость мышц (мышечная гипотония);
- дефекты развития костей таза;
- повышенная подвижность в суставах конечностей;
- характерное положение мизинца;
- недоразвитие ушных раковин;
- наличие «обезьяньей» складки;
- наличие кожной складки на шее.
После подтверждения диагноза могут быть назначены следующие методы исследования новорожденного:
- УЗИ брюшной полости;
- общий анализ крови и биохимический анализ крови;
- общий анализ мочи и биохимический анализ мочи;
- электрокардиография (ЭКГ);
- эхокардиография (ЭхоКГ);
- рентгенография.
Кроме того, рекомендуется в первые недели или месяцы после рождения пройти осмотр у следующих специалистов:
- оториноларинголог (ЛОР-врач);
- окулист;
- невропатолог;
- кардиолог;
- хирург;
- ортопед.
Заключение
В результате интенсивного изучения хромосом человека и хромосомных болезней на протяжении 35–40 лет сложилось учение о хромосомной патологии, которая имеет большое значение в современной медицине.
Список литературы
- Беляев Д.К. Общая биология. Базовый уровень. – 11 издание, стереотипное. – М.: Просвещение, 2012.
- Пасечник В.В., Каменский А.А., Криксунов Е.А. Общая биология, 10-11 класс. – М.: Дрофа, 2005.
- Агафонова И.Б., Захарова Е.Т., Сивоглазов В.И. Биология 10-11 класс. Общая биология. Базовый уровень. – 6-е изд., доп. – Дрофа, 2010.
Дополнительные рекомендованные ссылки на ресурсы сети Интернет
- Bibliofond.ru (Источник).
- Rh-conflict.narod.ru (Источник).
- Vse-pro-geny.ru (Источник).
Домашнее задание