Синдром франческетти: причины, симптомы, лечение
Содержание:
Лечение
В случае, когда заболевание диагностировано еще во время внутриутробного развития, женщине рекомендуют сделать аборт. В противном случае, сразу после рождения ребенку потребуется квалифицированное комплексное лечение по устранению дефектов, насколько это возможно. Терапия заключается в следующих действиях:
- Хирургическое вмешательство, в ходе которого устраняются дефекты лица. Обычно необходимо несколько операций, чтобы уменьшить проявления болезни. Все зависит от сложности случая.
- Стоматологические процедуры. Обычно у ребенка диагностируют не только проблемы с зубами, но и не правильный прикус, которые с помощью дантистов можно корректировать.
- Чтобы улучшить костнопроводимость звуков, потребуется установить слуховые аппараты.
Если у ребенка наблюдаются проблемы с дыханием и питанием, то потребуются операции по решению этих проблем (трахеотомия и гастростомия). Для дальнейшей адаптации ребенка потребуется работа с логопедами и сурдопедагогами.
Причины
СТК вызывается мутацией генов TCOF1, POLR1B, POLR1C или POLR1D. В случае мутации гена TCOF1 тип наследования является аутосомно-доминантным, хотя наблюдались очень редкие случаи аутосомно-рецессивных мутаций. Мутации POLR1B являются аутосомно-доминантными, тогда как при POLR1C они являются аутосомно-рецессивными, а мутации POLR1D могут быть аутосомно-доминантными или аутосомно-рецессивными.
Генетические заболевания определяются сочетанием генов для определенного признака, которые находятся на хромосомах, полученных от отца и матери. Доминантные генетические нарушения возникают, когда для появления заболевания необходима только одна копия ненормального гена. Для TCOF1, POLR1B и POLR1D аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации (спонтанного изменения гена) у пострадавшего человека. Примерно у 60% пациентов с синдромом Тричера-Коллинза мутация является новой, которая возникает случайно (спонтанно) без предшествующего семейного анамнеза заболевания (мутация de novo). Тем не менее, родители также могут быть слегка затронуты и не знать, что у них есть расстройство. Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50% при каждой беременности. Риск одинаков для детей мужского и женского пола. Независимо от того, наследуется ли мутация от матери или отца, похоже, что она не имеет отношения к тяжести состояния СТК у их детей.
Рецессивные генетические нарушения (например, СТК, вызванные мутациями POLR1C или POLR1D) возникают, когда человек наследует один и тот же аномальный ген для одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно не проявлять симптомов. Риск для двух родителей-носителей, чтобы передать оба дефектных гена и, следовательно, родить больного ребенка, составляет 25% при каждой беременности. Риск зачать ребенка, который будет носителем, как родители, составляет 50% при каждой беременности. Вероятность для ребенка получить нормальные гены от обоих родителей и быть генетически незатронутым для этой конкретной болезни составляет 25%.
Мутации гена TCOF1 вызывают большинство (приблизительно 80%) случаев синдрома Тричера-Коллинза. TCOF1 содержит инструкции, которые кодируют (создают) белок, известный как treacle. Точная роль, которую играет белок treacle в развитии СТК, полностью не понята. Исследователи определили, что treacle играет роль в создании определенных небольших структур в клетках, которые собирают белки (рибосомы)
Это особенно важно для формирования группы клеток, называемых клетками нервного гребня, которые образуются очень рано во время эмбрионального развития и дают начало большей части кости и хряща, лежащих под лицом
Состояния, которые возникают из-за дефектов в образовании (биогенезе) рибосом, называются рибосомопатиями. POLR1B кодирует субъединицу РНК-полимеразы 1, тогда как POLR1C и POLR1D кодируют субъединицы РНК-полимеразы I и III, каждая из которых также важна для биогенеза рибосом. Вероятно, что мутации в TCOF1, POLR1B, POLR1C и POLR1D вызывают недостаточную сборку белка и не позволяют определенным клеткам нервной системы и нервного гребня удовлетворять свои потребности в пролиферации и росте во время развития эмбриона. Поскольку СТК сильно различается, исследователи предполагают, что дополнительные генетические и, возможно, экологические факторы также могут играть роль в варьируемой степени тяжести заболевания. В поддержку этой концепции недавние экспериментальные данные показали, что белок treacle играет критическую роль в защите от вызванного окислительным стрессом повреждения ДНК в нервных клетках, а также в ориентации гребня во время деления нервных клеток, которые впоследствии влияют на развитие головы и лица.
Характеристики
Синдром Франческетти – серьезное врожденное расстройство черепно-лицевого развития, характеризующееся многочисленными аномалиями, которые ограничены головой и лицом. Гипоплазия костей лица, особенно челюстной и скуловой системы, является общей чертой.
Эта болезнь почти полностью отсекает ребенка от общества, делая его жизнь еще более тяжелой и болезненной.
Синдром Франческетти – это генетическое заболевание, вызванное мутацией генов, помогающих правильному развитию костей и лицевых тканей. Этот белок важен для развития лица как в утробе матери, так и после рождения. Мутация приводит к уменьшению его количества, изменяет клетки формирующие черты лица.
Нарушения в развитии костной ткани, вызывают сильную асимметрию, из-за чего слуховой и зрительный аппарат приобретают постоянные структурные патологии.
Диагностика
Стоит отметить, что выявление данного недуга происходит во время беременности. У сформировавшегося плода врачи диагностируют наличие синдрома Франческетти. Рекомендуется проводить исследование лицевой части с помощью биопсии. Однако этот способ не является безопасным, и поэтому врачи часто используют для этих целей УЗИ. Также необходимо у всех членов семьи взять анализы крови. После этого беременной женщине следует пройти фетоскопию. Заключительным этапом диагностики является взятие крови из плацентарных сосудов.
Причины синдрома Франческетти кроются в мутации генов, поэтому в зависимости от выраженности болезни врачи принимают решение о целесообразности дальнейших исследований. Если заболевание характеризуется экспрессивностью, то здесь сомнений быть не может. Однако в случае обнаружения некоторых отдельных симптомов специалисты обязаны продолжить наблюдение. Список мероприятий для выявления синдрома составляется врачом индивидуально, и в большинстве случаев это приносит свои плоды
Данное заболевание является довольно тяжёлым, и поэтому очень важно диагностировать его заранее, чтобы оставался шанс на исправление положения вещей
Признаки синдрома Тричера Коллинза
Интенсивность проявления генетического дефекта значительно варьируется. В медицине описаны основные симптомы заболевания, используемые для постановки диагноза:
- Недоразвитость челюстей – микрогнатия. У ребенка формируется очень узкий подбородок и маленький рот. Данные изменения могут сопровождаться нарушениями строения зубного ряда.
- Веки вследствие возникновения хромосомной аномалии приобретают треугольную форму. Такое состояние обозначается термином «коломба».
- Скуловые кости и орбиты глаз также уменьшены в размере. Это придает внешности пациентов специфический вид. Лицо кажется «затонувшим» за счет глубоко посаженных органов зрения и крупного носа.
- Многие дети рождаются с дефектом неба – «волчьей пастью». Данная аномалия приводит к затруднению приема пищи, поскольку происходит формирование канала между ротовой и носовой полостью.
- Значительно страдают органы слуха. Это проявляется тугоухостью, вплоть до полной глухоты. Нарушение связано с изменением нормального строения анализатора. Косточки, обеспечивающие восприятие звуков, деформированы, при этом даже осуществление операции не приводит к восстановлению слуха. У некоторых пациентов диагностируется недоразвитость или полное отсутствие ушных раковин.
- В ряде случаев синдром Тричера Коллинза сопряжен с опасными болезнями, например, пороками развития сердца. Тогда прогноз значительно ухудшается. У некоторых детей при рождении обнаруживается полное заращение носовых ходов (атрезия хоан), сопровождающееся респираторными нарушениями.
Согласно имеющейся статистике, лидирующую позицию по распространенности среди клинических проявлений синдрома Тричера Коллинза занимает недоразвитость или гипоплазия костей скулового комплекса, диагностируемая у 81% пациентов. При этом структуры нижней челюсти деформированы и уменьшены в 78% случаев. Подобные нарушения приводят к развитию таких проблем, как изменение прикуса. У большинства детей с патологией он открытый, что может затруднять прием пищи. 28% пациентов страдают от чрезмерно высокой небной дуги, в которой формируется расщелина. Данная патология носит название «волчья пасть».
Со стороны органов зрения у людей с синдромом Коллинза регистрируется уменьшение наклона пальпебральной щели. Такая аномалия диагностируется в 89% случаев и приводит к развитию офтальмологических симптомов. У 69% пациентов отмечается коломба век – дефект, при котором часть структуры отсутствует.
При рентгенографическом исследовании, а также применении КТ врачи выявляют многочисленные изменения со стороны органов слуха. Дети с синдромом рождаются со сращением молоточка, наковальни и стремечка. В ряде случаев костные структуры могут отсутствовать вообще. Подобные дефекты приводят к тугоухости или полной глухоте.
Источники
- https://simptom-lechenie.ru/sindrom-trichera-kollinza-foto-simptomy-i-lechenie.html
- https://vlanamed.com/sindrom-trichera-kollinza/
- https://baby-calendar.ru/vrozhdennye-poroki/sindrom-trichera-kollinza/
- https://womanadvice.ru/sindrom-trichera-kollinza-chto-takoe-chelyustno-licevoy-dizostoz-i-kak-mozhno-pomoch-malyshu
- https://dialogpress.ru/bolezni/sindrom-trichera-kollinza
- https://ProSindrom.ru/genetic/sindrom-trichera-kollinza.html
- https://sindrom.info/trichera-kollinza/
- https://www.KrasotaiMedicina.ru/diseases/genetic/Treacher-Collins-syndrome
Синдром Франческетти: причины,симптомы и лечение заболевания
Синдром Франческетти представляет собой аутосомно-доминантное заболевание, характеризующееся деформациями фасциально-лицевой области. Впервые он был описан в 1900 году английским офтальмологом Тричером Коллинзом.
Характеристика патологии
Заболевание характеризуется наличием у больного многочисленных лицевых уродств. Так как патология развивается в период внутриутробного развития, сразу после рождения можно установить синдром Тричера Коллинза–Франческетти.
Это заболевание имеет свои проявления и практически полностью отрезает ребенка от социума, делая его жизнь еще тяжелее и болезненнее. Нарушения развития костной ткани и окостенения вызывают сильную асимметрию лица, из-за которой слуховой и зрительный аппараты приобретают неизменные структурные патологии.
Синдром Франческетти – редчайшее генетическое заболевание, которое встречается у 1 из 50000 новорожденных. Пока медицине неподвластен контроль генетических мутаций, поэтому при наличии в роду любых патологий, необходимо обязательно посещать генетика, который поможет просчитать вероятность развития у будущего ребенка каких-либо генетических отклонений.
Причины возникновения патологии
Развитие заболевания связывают с нарушениями внутриутробного развития в период 6-7 недели. Нарушения локализуются в эмбриональном элементе первой жаберной дуги.
Синдром Франческетти может иметь различную степень выраженности симптомов: от незаметных до сильно выраженных патологий развития черепа. Синдром наследуется по аутосомно-доминантному признаку и имеет высокий процент проявления подобных фенотипических патологий у детей.
Признаки наличия синдрома
Внешне синдром проявляется косыми глазными щелями, внешние уголки которых опущены, иногда наблюдается колобома века (отсутствие части нижнего или верхнего века), врожденная катаракта, микрофтальм, парез мышц, ответственных за движение глаза.
В челюстно-лицевой системе наблюдается недоразвитие скуловой кости, верхне- и нижнечелюстной. В результате чего асимметрия лица значительная, иногда у больных присутствует волчья пасть, возможно выталкивание языка, способствующее преграждению ротоглотки и вызывающее заболевания дыхательных путей.
Зубы часто недоразвиты, широко поставлены, имеются проблемы с прикусом. Гипоплазия нижней части лица придает ему птичий вид. В редких случаях наблюдаются поражения крупных кровеносных стволов, сердца, отставание в развитии, внутренняя гидроцефалия.
Легкая степень синдрома практически не имеет структурных изменений лицевого отдела, при средней тяжести – перечисленная симптоматика выражается выборочно.
Тяжелая степень синдрома характеризуется полным отсутствием выделенных черт лица у ребенка.
По статистическим данным, синдром средней тяжести проявляется чаще всего, но в большинстве случаев устранить дефекты удается при помощи пластической хирургии.
Через гастростому (образованное соустье) осуществляется питание и дыхание пациента. Улучшить внешность можно при помощи пластической операции, но выполняется она по желанию и только по достижении определенного возраста.
Хирургическое изменение внешности требует постепенности и ступенчатого лечения. Дефекты могут устраняться за несколько лет, и даже десятилетия. В некоторых случаях невозможно полное исправление деформаций лица, поэтому врачи путем хирургического вмешательства могут лишь незначительно снизить проявление симптомов и улучшить качество жизни больного.
Попытки восстановить слух путем хирургического вмешательства (для исправления структуры слуховых косточек) не оказали на больных положительного эффекта, поэтому лучше для этой цели использовать слуховые аппараты, подобранные с учётом индивидуальных патологий строения внутреннего и среднего уха.
Синдром Франческетти: фото больных
На фото видно, что люди с синдромом Тричера Коллинза ведут не менее активную жизнь, чем здоровые люди. Они учатся воспринимать себя, любить свое лицо, стремятся помочь таким же детям, которые не имеют желания полонценно жить и боятся выйти на улицу из-за насмешек других детей.
Даже у абсолютно здоровых родителей может появиться ребенок с генетической патологией.
На фото до и после пластической операции у девочки видна значительная разница. Поэтому не стоит отчаиваться и раньше времени складывать руки. Всегда можно найти способ улучшить свою внешность.
Синдром Фрейли у детей: лечение
Несмотря на то, что синдром Фрейли считается врождённым заболеванием, существуют способы его эффективного лечения. Болезнь перейдёт в хроническую форму и будет продолжать прогрессировать, если ограничиться только борьбой с симптомами. Чтобы вернуть человека к здоровому образу жизни используют хирургическое вмешательство.
Для пациентов детского возраста применяют консервативное лечение медицинскими препаратами. Такими лекарственными средствами, способными облегчить недуг считаются:
- Даприл — стабилизирует артериальное давление, способствует восстановлению кровяного снабжения почек.
- Рениприл — нормализует повышенное давление в сосудах, усиливает выведение мочи из организма;Витамины групп А, С, Д.
- Уролесан, Канефрон — препараты, которые предотвращают образование камней и способствуют их выведению.
Для профилактики развития повторного обострения необходимо вести правильный образ жизни, отказаться от вредных привычек, заботиться о питании и соблюдении диеты.
Лечение синдрома Фрейли у детей
- Только после проведения всех необходимых видов диагностики и тщательного их анализа врач может назначить адекватное лечение.
- Лечить сам синдром Фрейли препаратами нет необходимости, да и возможности таковой нет.
- Все профилактические и лекарственные мероприятия назначаются лишь для устранения болезненной симптоматики и сопутствующего заболевания.
- В ходе исследований урологу также придется проанализировать риски передавливания почечной лоханки.
- Если оказываемое давление является несущественным и не приводит к каким-либо последствиям, его можно не трогать, а попросту наблюдать за ним — в некоторых случаях человек живет всю жизнь с синдромом Фрейли, так ни разу и не столкнувшись с проблемами мочевыделительной системы.
- Если же положение является критичным, то доктор может принять решение задействовать единственный действенный способ коррекции отростков верхней почечной артерии — хирургическое вмешательство.
- Операция проводится лишь в тех случаях, когда у больного все в порядке со здоровьем, и ему позволяет возраст.
Оперативное лечение синдрома Фрейли К современным хирургическим методам решения проблемы синдрома Фрейли относятся:
- инфундибулопластика — операция по увеличению размеров чашечно-лоханочного устья;
- инфундибулоанастомоз — операция по перемещению артериального ответвления;
- инфундибулопиелонеостомия — операция по перемещению артериального ответвления в специально созданный путь между почечными лоханкой и чашечкой;
- каликопиелонеостомия — аналог инфундибулопиелонеостомии;
- интрарентальная вазопексия — операция по разведению путей патологически расположенного артериального ответвления и мочевых протоков.
Лечение синдрома Фрейли лекарствами
- Что касается препаратного лечения синдрома Фрейли, то все, назначенные доктором, средства будут направлены лишь на подавление острой симптоматики и облегчение состояния больного.
- Для понижения почечного давления чаще всего назначают гипотензивные лекарственные средства: Даприл, Каптоприл, Рениприл, Эналаприл, Каптопрес, Фоззиноприл и т.д.
- Для устранения болевого синдрома больные могут принимать спазмолитики (Но-шпа, Спазмалгон, Папаверин, Спазмил и т.д.) и болеутоляющие комплексного действия (Бралангин, Спазмалин, Реналган, Новиган, Баралгин и т.д.).
Синдром Тричера Коллинза причины
Синдром Тричера Коллинза является аутосомно — доминантным заболеванием. Это значит, что дефектный ген этой болезни не связан с половой хромосомой, а значит, равносильно может появиться и у мужчин и у женщин. Кроме того, этот ген является доминантным, что означает, что при его наличии в организме, синдром Тричера Коллинза проявится в 100% случаев наличия этого гена.
Таким образом, развитие заболевания Тричера Коллинза не зависит от воздействия каких-либо вредных внешних и внутренних факторов. Можно сказать, что это заболевание уже заложено в генетический код будущего ребенка и начинает раскрываться задолго до его рождения.
Причина появления синдрома Тричера Коллинза у новорожденного ребенка кроется еще на этапе эмбриогенеза плода и процесса закладки органов. Именно в это время, а точнее на 7-й неделе развития эмбриона, происходит мутация в определенной хромосоме человеческого генетического кода – 5-й хромосоме. Эта хромосома является самой длинной в геноме человека, и именно она отвечает за синтез материала для будущего костного скелета. В результате в этой хромосоме происходит особая мутация — так называемая «нонсенс-мутация». Особенность этой мутации кроется в особенностях внутриклеточного синтеза белка. В норме такой важный процесс как биосинтез белка происходит следующим образом: цепь ДНК «перезаписывает» информацию на вспомогательную единицу – РНК. Буквально говоря РНК клонируется, записывая на себя определенную последовательность участков ДНК. Эти участки представляют собой определенную последовательность составных частей нуклеотидов, каждый из которых несет свою особую информацию. Но кроме нуклеотидов существуют и особые гены, которые получили название «стоп-кодонов». Эти гены выполняют особую функцию — в последующей сборке белка на основе РНК они в заканчивают постройку молекулы белка.
После того, как будет создана РНК с полной информацией, аналогичной информации материнской ДНК, она транспортируется в особый клеточный орган-рибосому. Именно она занимается синтезированием будущего белка — основы клеточной структуры определенных органов. РНК проходит сквозь рибосому, а точнее через ее функциональные участки. Эти участки взаимодействуют с РНК, «считывают» с нее информацию и вырабатывают белковые цепи, где каждая из них соответствует своему нуклеотиду на РНК. Но когда функциональный центр взаимодействует с описанным выше геном стоп кодоном, то он получает информацию о прекращении синтеза белка. Говоря своими словами, стоп кодоны, как бы «отрезают» от общей массы отдельные полипептидные цепи, где каждая имеет свою структуру. Позже эти цепи будут собраны в белковые молекулы.
Но при синдроме Тричера Коллинза в 5-й хромосоме, в ее гене под названием TCOF1 происходит сбой — на месте нормальных нуклеотидов, способных создать полипептидную цепь, образуется стоп-кодон. В результате, при дальнейшем синтезе происходит преждевременное окончание сборки белка, и такой белок получается дефектным. Как результат, развивается синдром гаплонедостаточности — количества образуемого белка при такой недостаточности просто недостаточно для синтезирования будущего прототипа костной структуры лицевой части черепа и последующего правильного развития из него самой костной структуры.
Как следствие этого, развивается формирование целого ряда деформаций лицевой части черепа: нарушение пропорций ее костной части, атрезия (недоразвитие) ушных раковин и наружного слухового прохода, полное или частичное нарушение формирования правильных черт лица.
Медицинские новости
Специалисты заявляют о нестабильной эпидемиологической ситуации по заболеваемости коклюшем в различных регионах РФ – в том числе, и в Санкт-Петербурге. По данным Федеральной службы по надзору…
Мигрень широко распространена во всем мире, изучена лучше других типов головной боли и является второй ведущей причиной потерянных лет жизни . На сегодняшний день в России от этого заболевания страдают более 20 миллионов человек. При этом большинство из них не знают о своем диагнозе…
Фотовыставка «Видеть главное», посвященная пациентам с псориазом, открылась на портале МБОО «Кожные и аллергические болезни» в виртуальном формате. «Видеть главное» — это 12 портретов, выполненных в технике стерео-варио, которая позволяет увидеть фото со следами псориаза и без них в зависимости от того, под каким углом смотрит посетитель.
Статистика показывает, что ишемическая болезнь сердца и инсульт уносят больше всего человеческих жизней во всем мире. Коронавирус COVID-19 — серьезное явление, но про здоровье других органов в тоже время забывать не стоит. На здоровье сердца влияет не так много факторов…
Александровскую больницу закрыли на карантин по постановлению главного санитарного врача Санкт-Петербурга. Это связано с тем, что в этой больнице умер 55-летний пациент с подтвержденным коронавирусом Covid-2019
Причины
Развитие заболевания связано с нарушениями внутриутробного развития на 6-7 неделе.
Расстройство возникает из-за мутаций гена TCOF1 или POLR1C, POLR1D. Это аутосомно-доминантное состояние, что означает, что одна копия измененного гена в каждой клетке достаточна, чтобы вызвать расстройство.
Около 60 процентов случаев являются результатом новых мутаций и встречаются у людей, не имеющих истории семейного расстройства. Чаще, человек наследует измененный ген от пострадавшего родителя.
Когда синдром Франческетти вызывается мутациями гена POLR1C, состояние имеет аутосомно-рецессивный характер наследования. Такое наследование означает, что обе копии гена в каждой клетке имеют мутации.
Родители индивидуума с аутосомно-рецессивным состоянием имеют одну копию мутированного гена, но обычно не проявляют признаков и симптомов состояния.
Мутации гена TCOF1 являются наиболее распространенной причиной расстройства, составляя от 81 до 93 процентов всех случаев.
Белки, полученные из генов TCOF1, POLR1C и POLR1D, играют важную роль в раннем развитии костей и других тканей лица. Они участвуют в получении молекулы, называемой рибосомальной РНК (рРНК). Рибосомальная РНК помогает собрать белковые строительные блоки (аминокислоты) в новые белки, которые необходимы для нормального функционирования и выживания клеток.
Мутации TCOF1, POLR1C, POLR1D уменьшают продукцию рРНК. Исследователи предполагают, что уменьшение количества рРНК может спровоцировать саморазрушение (апоптоз) определенных клеток, участвующих в развитии костей и тканей лица.
Аномальная гибель клеток приводит к специфическим проблемам развития лица, обнаруженным при синдроме Франческетти.
Симптоматика
Синдром характеризуется полиморфностью клинических проявлений. Больных с СТК можно узнать сразу. Такие дети имеют характерный внешний вид и часто похожи друг на друга.
Клинические признаки синдрома:
- нарушение нормальной формы глазной щели, широкий разрез глаз и опущение их внешнего края, антимонголоидные глаза;
- гипоплазия скуловых костей и надбровных дуг;
- асимметрия лица;
- непропорционально крупный нос;
- вдавленное внутрь лицо;
- мелкий подбородок,
- незаращение твердого неба;
- расщелина верхней губы;
- рост волос на щеках;
- поражение органа слуха — недоразвитие слуховых косточек, барабанной полости и ушной раковины; атрезия слухового прохода; тугоухость; предкозелковые фистулы;
- поражение полости рта — «готическое» небо; фарингеальная гипоплазия — сужение глотки и дыхательных путей; «открытый», неправильный прикус; ограничение возможности открытия рта различной степени тяжести; отсутствие зубов; смещение языка назад с перекрытием дыхательных и пищеварительных путей; дефект мягких тканей ротовой полости;
- поражение органа зрения — колобома нижнего века, отсутствие ресниц; косоглазие, снижение остроты зрения;
- деформированные большие пальцы рук.
Деформация лица и черепа может сочетаться с пороками развития внутренних органов: сердца, позвоночника, слухового анализатора, желез внешней и внутренней секреции, дыхательных путей. У больных детей нарушается адаптация в обществе. Они стесняются окружающих и избегают контактов с ними. Это приводит к формированию комплекса неполноценности и развитию депрессии. При этом интеллект полностью сохраняется: больные адекватно воспринимают информацию и правильно развиваются в моральном и физическом плане.
Клинические признаки синдрома имеют различную степень выраженности: от малозаметных деформаций до тяжелых уродств, при которых полностью стерты черты лица. У больных в запущенных случаях появляются проблемы с жеванием и глотанием, произношением отдельных звуков, зрением и слухом.
Стадии синдрома определяются сложностью мутационного процесса и интенсивностью клинических признаков:
СТК тяжелой степени
- Начальная стадия характеризуется практически незаметными изменениями на лице. Больные дети ни чем не отличаются от здоровых и ведут нормальный образ жизни.
- Средняя стадия проявляется всеми перечисленными выше нарушениями. Аномальная деформация лицевых костей достаточно сильная. Возможны трудности с дыханием, принятием пищи, нарушение слуха, проблемы с зубами.
- Тяжелая стадия — полное отсутствие лица, невозможность рассмотреть его черты. Даже пластическая хирургия не может помочь больным.
Диагностика
Врачи-кардиологи выслушивают жалобы больных, собирают анамнез жизни и болезни, выясняют, какие сердечные заболевания перенес пациент. Затем переходят к физикальному осмотру и инструментальному обследованию.
Основным диагностическим методом является электрокардиография. Характерные ЭКГ-признаки:
-
отсутствие зубца Р,
- признаки фибрилляции – мелкие и частые волны ff,
- признаки трепетания предсердий – крупные и редкие волны FF,
- ритм желудочков несинусового происхождения,
- изменение комплексов QRS,
- постоянные интервалы R — R, Р-Р,
- количество сердечных сокращений в минуту не превышает 40-60 раз,
- полное разобщение предсердного и желудочкового ритмов,
- регулярный желудочковый ритм.
пример: ЭКГ пациента, 80 л., синдром Фредерика ( фибрилляция предсердий + АВ-блокада 3 ст.) . Ритм не синусовый, правильный. Замещающий ритм – узловой, с блокадой правой ножки пучка Гиса, ЧСС – 32 уд\мин (фото: therapy.odmu.edu.ua)
Среди дополнительных методов диагностики наиболее информативными являются: холтеровское мониторирование ЭКГ и эхокардиография. Первый метод позволяет регистрировать данные электрокардиографии в течение суток независимо от состояния и активности больного, а второй выявляет имеющиеся морфологические дефекты в структуре сердца.
Челюстно-лицевой дизостоз – симптомы
Выявить челюстно-лицевой дизостоз можно уже при первом осмотре пациента:
- У таких детей отмечается выраженная недоразвитость скуловых костей, челюсти.
- Ушные раковины полностью не сформированы: уши у ребенка имеют малый размер, а слуховой канал недоразвит.
- Разрез глаз заметно сужается.
- Тип и выраженность нарушения может варьироваться, однако всегда происходит сбой в работе слухового аппарата, дыхательной системы.
Самым ярким симптом, сопровождающим болезнь Тричера-Коллинза, является изменение нормальной формы глаз. Глазная щель сужается, в результате чего глазные яблоки опускаются. Возможны и другие проявления синдрома Тричера-Коллинза:
- дефект мягкой ткани ротовой полости;
- впалый подбородок;
- нарушение слуха;
- нарушение прикуса;
- расщепление верхнего неба;
- изменение прикуса.
Синдром Тричера-Коллинза – степени
Как отмечалось выше, патология развивается постепенно, прогрессируя со временем. Синдром Тричера-Коллинза в начальной степени характеризуется незначительной гипоплазией лицевых костей. Диагностируется уменьшение в размерах скуловых костей, из-за чего лицо выглядит слегка вытянутым. Такое изменение именуют первой степенью заболевания.
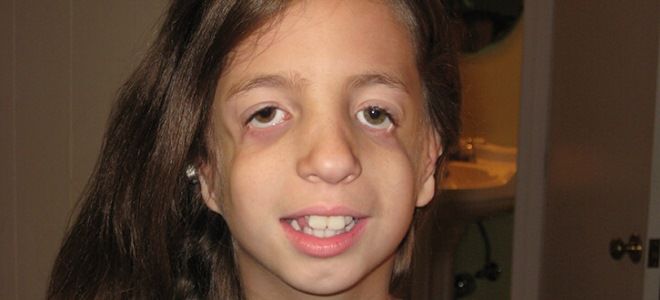
Челюстно-лицевой дизостоз или синдром Тричера-Коллинза второй степени сопровождается недоразвитием слуховых проходов. Нижняя челюсть значительно меньшего размера, глазная щель сужается, что приводит к нарушению зрения. При тяжелой форме лицо практически отсутствует. Нос, скуловые кости, верхняя и нижняя челюсти деформируются настолько, что пациент становится неузнаваемым для окружающих.
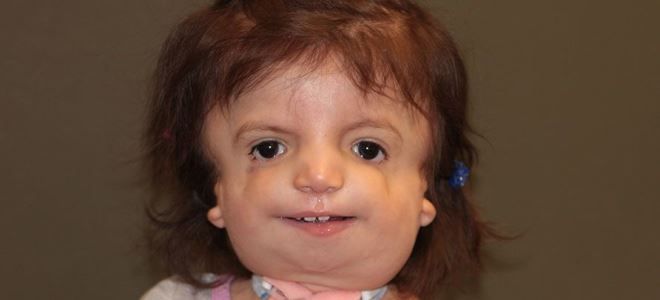
Жизнь с мутацией
Та самая Джуллиана, которая страдает от этого синдрома в самом тяжелом его проявлении. С точки зрения развития, такие люди абсолютно ничем не отличаются от среднестатистического человека, но при определенных осложнениях могут быть проблемы с дыханием или питанием В Великобритании проживает молодой человек – Джон Ланкастер, который, несмотря на это серьезное заболевание, продолжает вести полноценную жизнь. На данный момент ему около 26 лет. Сразу после рождения родители отказались от нездорового ребенка, испугавшись предстоящих трудностей, связанных с его воспитанием. Несмотря на это, ребенку были сделаны сложные операции, в результате которых его внешность немного изменилась. На данный момент молодой человек активно помогает и поддерживает детей, страдающих от синдрома Тричера Коллинза, а также путешествует по миру. На примере своей жизни он доказывает, что несмотря на генетические аномалии нужно жить и радоваться каждому моменту. У Джона Ланкастера есть любящая девушка, которая его во всем поддерживает.
Младшая сестра Аделины Сотниковой (российской фигуристки и олимпийской чемпионки) – Мария также имеет генетическое заболевание. Еще в роддоме, когда стало известно о болезни младенца, врачи стали рекомендовать оставить его. Но родители не отказались от дочери и решили заботится о ней. Маша Сотникова пережила уже достаточно много операций, которые скорректировали ее внешность. Но в связи с тем, что девочка растет, ей предстоят еще хирургические вмешательства. Аделина Сотникова все свои гонорары тратит на ее лечение и никаким образом не стесняется своей сестры. Она считает, что таких людей нужно воспринимать, как особенных и требующих особую заботу.
Джулиана – это самый сложный и ужасный случай проявления болезни. Родилась девочка в 2003 году практически без лица, так как около 40% лицевых костей отсутствовала. Но несмотря на это ребенок выжил и после десятка перенесенных операций ведет обычную жизнь ребенка. Постепенно из года в год, с каждой новой процедурой, она обретает лицо. Джулиана вскоре пойдет в школу и будет вести жизнь, которая характерна для всех остальных детей.